
Projetos
Electronic and ionic transport of ions in water confined
in carbon nanotubes: a combined first principles
calculations
Executor:
PhD student: Alexsandro Kirch
Supervisor:
Prof. Dr. Caetano Rodrigues Miranda

The unusual physical properties of confined fluids at nanoscale can play an important role in a plethora of chemical, geochemical and environmental processes. Nowadays, there is a strong interest on these systems for developing devices for nanofluidics applications. Through external electric field, it was shown that it is possible to modulate ionic conductivity, promoting nanofluidic systems as a logical transistor (Nano letters, v. 15, n. 4, p. 2365-2371, 2015). The ability to control the flow of ions through the nanochannel has several technological applications. Despite of this critical significance, the structure and properties of these confined fluids are still unclear, in particular for the case of electrolytes in aqueous solutions. Here, we combine first-principles electronic transport calculations with ab initio molecular dynamics (AIMD) to determine the electronic and ionic transport of ions through an aqueous solution confined at carbon nanotubes (CNT). Using classical and first principles molecular dynamics, we have investigated the ions (H, Li, Na and Cl) in aqueous solution flow within (6,6) metallic CNTs. To access the effects of the ions on the CNT electronic transmittances, calculations using non-equilibriums Greens Functions methodology has been performed based on snapshots from MD calculations using the Transampa and Siesta codes. The AIMD and ionic relaxations were performed using the Quantum Expresso package within the Density Functional Theory with revised PBE functional (revPBE) with van der Waals density functional correction (vdW-DF). The results suggest that Li, Cl, Na and H insertion in the Carbon nanotubes lead to different levels of doping, namely, a Dirac cone energy displacement with respect to the different chemical species. Due to size constrains, the (6,6) CNT allows only the flow of a single line of atoms or molecules, and thus, the ion is not completely solvated. However, even in the presence of water, the influence of these ions on the carbon nanotubes electronic properties has shown to be significant in the Dirac cone displacement. Based on state-of-art first principles calculations, we have been able to characterize the electronic (current and transmittance) and ionic current of confined fluids (electrolytic aqueous solution) with CNT. In particular, an electronic current signature of distinct ions can been observed, which could lead to potential applications as sensors and nanofluidic devices.
Determinação das interações fluído-fluído e fluído-rocha
sob condições de baixa salinidade através
de simulações moleculares
Executor:
PhD student: Oscar Andres Babilonia Perez
Supervisor:
Prof. Dr. Caetano Rodrigues Miranda
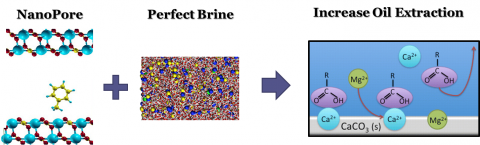
Com a escassez de novas reservas, aumento da demanda e escalada de preços de derivados do petróleo, processos de recuperação melhorada de petróleo (EOR) tornaram-se não apenas necessários, mas economicamente viáveis. Esses processos visam revitalizar reservatórios maduros, aumentando a produção e estendendo assim seus ciclos de vida. O ponto chave nesses processos de EOR é a diminuição da tensão interfacial e controle da viscosidade do óleo. Nesse contexto, diversas técnicas promissoras vêm sendo propostas. Dentre elas, destacamos o uso injeção de salmoura com baixa salinidade (Lo-sal ou “smart water”). Com a injeção de água com baixa salinidade (LoSal) verificou-se que a composição e presença de íons na água levam a um aumento significativo na recuperação. Diversos grupos, inclusive o nosso, vêm estudando a interação entre Ca2 +, Mg2+ e íons SO4_2+ e seus efeitos sobre o processo de EOR. A partir dessas observações, alguns mecanismos foram propostos para explicar a dessorção dos componentes do óleo por essa injeção de água de baixa salinidade. Entretanto, esses mecanismos não são completamente entendidos e variam de acordo com o ambiente físico-químico e eletrostático encontrado nos reservatórios, bem como detalhes das interações fluido-fluido e fluido-rocha. A compreensão desses mecanismos e a formulação de uma solução salina ótima poderiam contribuir para proposição de novas soluções que levem ao aumento na produção.
Noncontact AFM First Principles Simulations of Functionalized
Silicon Tips on the Montmorillonite (001) Surface
Executor:
Pos Doc student: Raphael da Silva Alvim
Supervisor:
Prof. Dr. Caetano Rodrigues Miranda
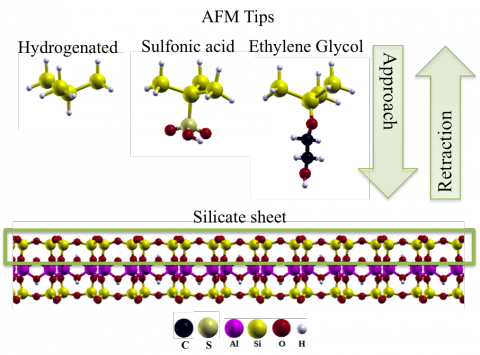
The aim of research is in solid state physics through the Quantum-ESPRESSO package. The first principles calculations are in density-functional theory level (DFT) with inclusion of van der Waals interactions. Particularly, the simulations are related to the interaction between organic molecules (like hydrocarbons and asphaltenes) and mineral surfaces (like montmorillonite and calcite), involving besides microscopic and spectroscopic analysis, the study of the influence of a continuum dielectric solvation model.
Simulation of adsorption and oxidation of aromatic
hydrocarbons on mineral surfaces
Executor:
Por definir
Supervisor:
Prof. Dr. Caetano Rodrigues Miranda
Carbonaceous-oil interfaces play a key role for most petroleum exploration, as the crude oil components affect the wettability of the mineral surface due the solubility in watery and oil mediums. However, these processes are far from complete understanding. Hence, to improve the Enhanced Oil Recovery on mineral surface the investigation of the interfacial effects and wetting properties of the mineral surfaces at a wide range of scale (from nano and to micro) are needed. For that a multiscale approach can be considered as an important tool for those investigations, which is based on the combination of first principles calculations, molecular dynamics and Lattice Boltzmann method (LBM). In this posdoc project, first of all, a fundamental understanding of the interaction between the hydrocarbon molecules, as the principal oil components, and the mineral surface is intended to be achieved. For that we will systematically investigate energetic, structural and electronic properties of a number of aromatic hydrocarbons, such as toluene (C6H5CH3), phenol (C6H5OH), aniline (C6H5NH2), naphthalene (C10H8), anthracene (C14H10) and asphaltene on the mineral surfaces, which present calcite (CaCO3), dolomite (Ca(Mg(CO3)2), quartz (SiO2), and muscovite (Al2Si4O10(OH)2) surfaces employing first-principles density functional theory (DFT) calculations. To carefully investigate the interaction mechanism between the hydrocarbons and mineral molecules, several analyses, such as charge redistribution and densities of states will be applied using the obtained lowest energy structures. Thus, the results systematically obtained in the project will help in achieving fundamental understanding of the interaction between aromatic hydrocarbon molecules with different functionalized groups and mineral surfaces at nanoscale.