|
xxxx
|
 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
Reproducibility in density functional theory calculations of solids
BaC6
Marília Junqueira Caldas
Tuesday, April 19, 2016
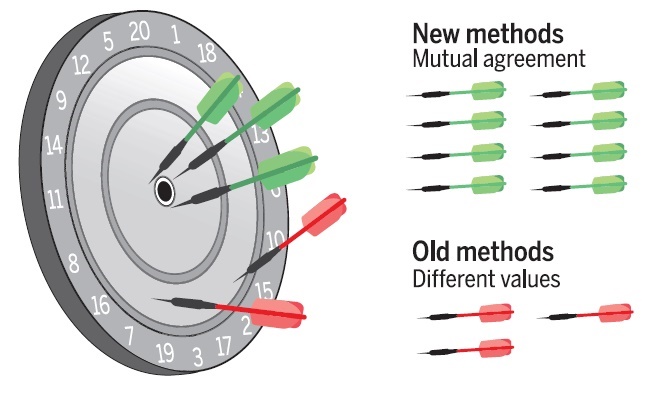
The widespread popularity of density functional theory has given rise
to an extensive range of dedicated codes for predicting molecular and
crystalline properties. However, each code implements the formalism in
a different way, raising questions about the reproducibility of such
predictions. We report the results of a community-wide effort that
compared 15 solid-state codes, using 40 different potentials or basis
set types, to assess the quality of the Perdew-Burke-Ernzerhof
equations of state for 71 elemental crystals. We conclude that
predictions from recent codes and pseudopotentials agree very well,
with pairwise differences that are comparable to those between
different high-precision experiments. Older methods, however, have
less precise agreement. Our benchmark provides a framework for users
and developers to document the precision of new applications and
methodological improvements.
View full text
|


